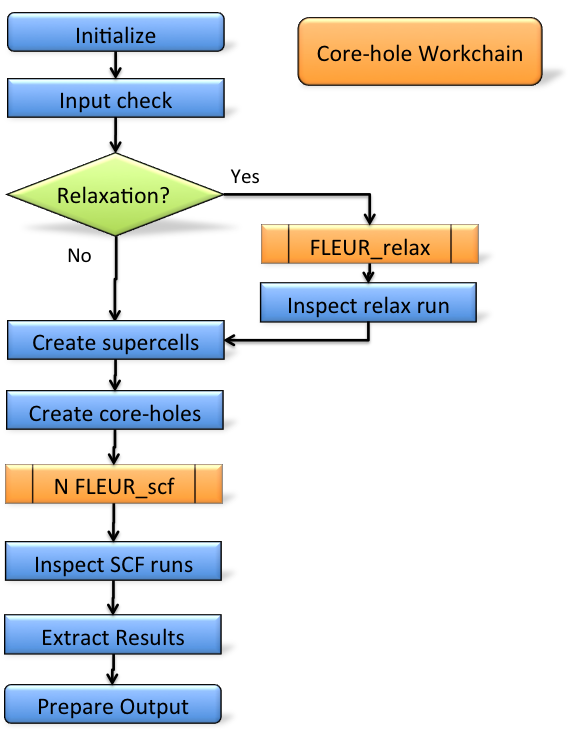
The core-hole workflow can be deployed to calculate absolute core-level binding energies.
Such core-hole calculations are performed through a super-cell setup.
The workflow allows for arbitrary corehole charges and of valence and charged type core-holes.
From a computational cost perspective it may be cheaper to calculate all relative initial-state
shifts of a structure and then launch one core-hole calculation on the structure to get an
absolute reference energy instead of performing expensive core-hole calculations
for all atom-types in the structure.
The core-hole workflow implements the usual FLEUR workflow interface with a workflow
control parameter node.
wf_parameters: Dict - Settings of the workflow behavior. All possible
keys and their defaults are listed below:
# -*- coding: utf-8 -*-'method':'valence',# what method to use, default for valence to highest open shell'hole_charge':1.0,# what is the charge of the corehole? 0<1.0'atoms':['all'],# coreholes on what atoms, positions or index for list,# or element ['Be', (0.0, 0.5, 0.334), 3]'corelevel':['all'],# coreholes on which corelevels [ 'Be1s', 'W4f', 'Oall'...]'supercell_size':[2,1,1],# size of the supercell [nx,ny,nz]'para_group':None,# use parameter nodes from a parameter group'relax':False,# relax the unit cell first?'relax_mode':'Fleur',# what releaxation do you want'relax_para':'default',# parameter dict for the relaxation'scf_para':'default',# wf parameter dict for the scfs'same_para':True,# enforce the same atom parameter/cutoffs on the corehole calc and ref'magnetic':True# jspins=2, makes a difference for coreholes
output_corehole_wc_para (Dict): Information of workchain results
More details:
output_corehole_wc_para: Dict - Main results of the workchain. Contains
Binding energies, band gaps, core-levels, atom-type information,
errors, warnings, other information. An example:
Currently there is no visualization directly implemented for plot fleur.
Through there in masci-tools there are methods to visualize spectra and binding energies