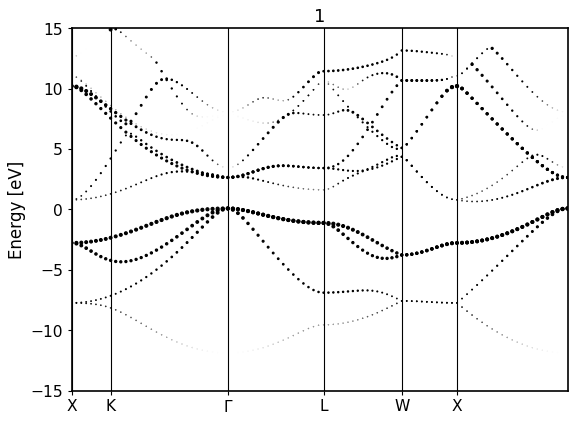
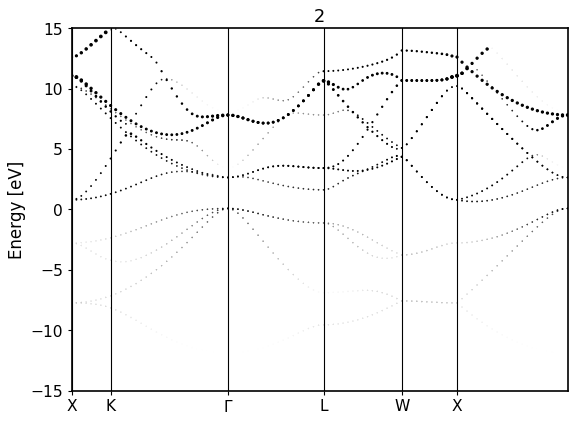
Calculates an electronic band structure on top of a given Fleur calculation (converged or not). It can be started from the crystal structure utilizing the FleurSCFWorkchain as a subworkchain
This workflow prepares/changes the Fleur input with respect to the kpoint set and bandstructure/DOS related parameters and manages one Fleur calculation.
Only the fleur input is required. However, it does not mean that it is enough to specify fleur
only. One must keep one of the supported input configurations described in the
Supported input configurations section.
wf_parameters: Dict - Settings of the workflow behavior. All possible
keys and their defaults are listed below:
# -*- coding: utf-8 -*-'mode':'band','kpath':'auto',#seek (aiida), fleur (only Max4) or string to pass to ase'klistname':'path-3','kpoints_number':None,'kpoints_distance':None,'kpoints_explicit':None,#dictionary containing a list of kpoints, weights#and additional arguments to pass to set_kpointlist'sigma':0.005,'emin':-0.50,'emax':0.90,'add_comp_para':{'only_even_MPI':False,'max_queue_nodes':20,'max_queue_wallclock_sec':86400},'inpxml_changes':[],
mode is a string (either band``(default)or``dos). Determines, whether a bandstructure or density of states calculation is performed. This sets the band and dos switches in the output section of the input file accordingly.
kpath is only used if mode='band' to determine the kpath to use. There are 5 different options here:
auto Will use the default bandpath in fleur for both Max4 or Max5. If klistname is given the corresponding kpoint path is used for Max5 version or later
A dictionary specifying the special points and their coordinates. Only available for versions before Max5. Will generate a kpath with kpoints_number points
This functionality only works for standardized primitive unit cells.
skip nothing is done
all other strings are used to generate a k-path using bandpath() for example GMKGALHA. This option supports both kpoints_number and kpoints_distance for specifying the number of points
kpoints_number integer specifying the number of kpoints in the k-path (depending on the kpath option)
kpoints_distance float specifying the distance between kpoints in the k-path (depending on the kpath option)
kpoints_explicit dictionary, which is used to create a new kpointlist in the input. The dictionary is unpacked and used as the argument for the set_kpointlist() function
klistname str, will be used to switch the used kPointList for fleur versions after Max5 (if kpath='auto' or mode='dos')
sigma, emin, emax floats specifying the energy grid for DOS calculations
The FleurBandDosWorkChain workchain has several
input combinations that implicitly define the workchain layout. Only scf, fleurinp and
remote nodes control the behaviour, other input nodes are truly optional.
Depending on the setup of the given inputs, one of three supported scenarios will happen:
scf:
SCF workchain will be submitted to converge the charge density which will
be followed by the bandsturcture or DOS calculation. Depending on the inputs given in the SCF
namespace, SCF will start from the structure or FleurinpData or will continue
converging from the given remote_data (see details in SCF WorkChain).
remote:
Files which belong to the remote will be used for the direct submission of the band/DOS
calculation. inp.xml file will be converted to FleurinpData and the charge density
will be used as the charge density used in this calculation.
remote + fleurinp:
Charge density which belongs to remote will be used as the charge density used in the
band/DOS calculation, however the inp.xml from the remote will be ignored. Instead, the given fleurinp will be used.
The aforementioned input files will be used for direct submission of the band/DOS
calculation.
Other combinations of the input nodes scf, fleurinp and remote are forbidden.
Warning
One must follow one of the supported input configurations. To protect a user from the
workchain misbehaviour, an error will be thrown if one specifies e.g. both scf and remote
inputs because in this case the intention of the user is not clear either he/she wants to
converge a new charge density or use the given one.
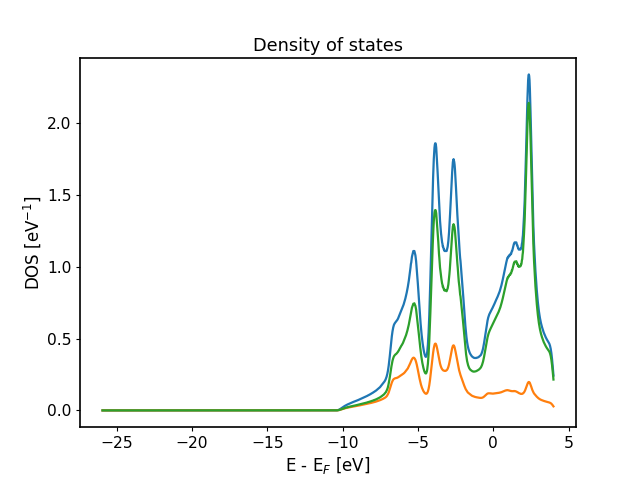
fromaiida_fleur.tools.plotimportplot_fleur# DOS calcplot_fleur(76867)
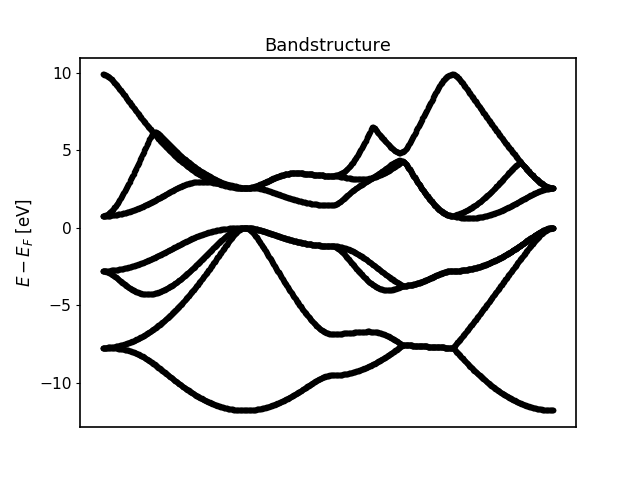
For the bandstructure visualization it depends on the File produced.
Old bandstructure file:
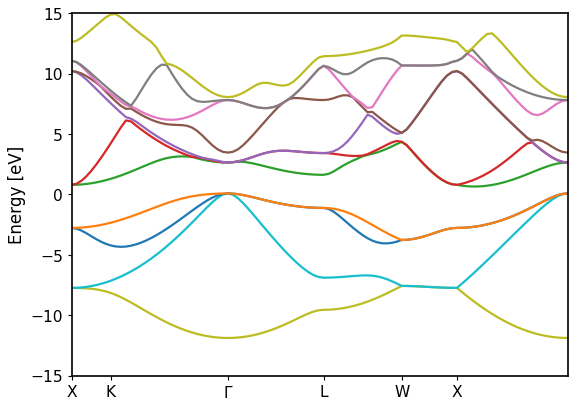
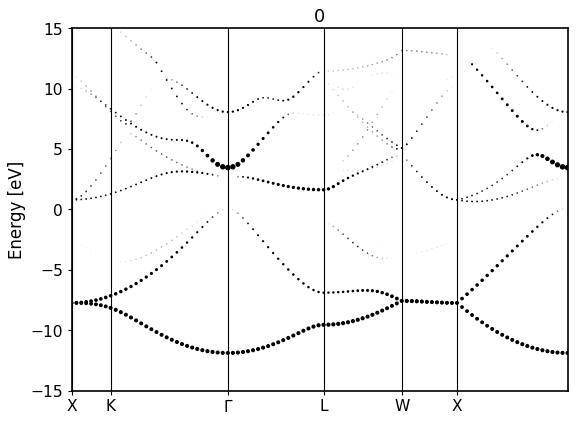
Bandstructure `band_dos.hdf` file with l-like charge information:
Band resolved bandstructure and fat-bands for the different channels.
Spin and combinded DOS plus band structure visualizations are in progress…
Multi node just does a bunch of single plots for now.
# -*- coding: utf-8 -*-fromaiida_fleur.workflows.banddosimportFleurBandDosWorkChainfromaiida.ormimportDict,load_nodefromaiida.engineimportsubmitfleur_code=load_node(FLEUR_PK)inpgen_code=load_node(INPGEN_PK)structure=load_node(STRUCTURE_PK)wf_para=Dict(dict={'mode':'band','kpath':'auto',#seek (aiida), fleur (only Max4) or string to pass to ase'klistname':'path-3','kpoints_number':None,'kpoints_distance':None,'kpoints_explicit':None,#dictionary containing a list of kpoints, weights#and additional arguments to pass to set_kpointlist'sigma':0.005,'emin':-0.50,'emax':0.90,'add_comp_para':{'only_even_MPI':False,'max_queue_nodes':20,'max_queue_wallclock_sec':86400},'inpxml_changes':[],})wf_para_scf=Dict(dict={'fleur_runmax':3,'density_converged':0.001,'mode':'density','itmax_per_run':30,'add_comp_para':{'only_even_MPI':False,'max_queue_nodes':20,'max_queue_wallclock_sec':86400}})options=Dict(dict={'resources':{'num_machines':1,'num_mpiprocs_per_machine':2},'withmpi':True,'max_wallclock_seconds':600})options_scf=Dict(dict={'resources':{'num_machines':1,'num_mpiprocs_per_machine':2},'withmpi':True,'max_wallclock_seconds':600})calc_parameters=Dict(dict={'kpt':{'nkpts':500,'path':'default'}})inputs={'scf':{'wf_parameters':wf_para_scf,'structure':structure,'calc_parameters':calc_parameters,'options':options_scf,'inpgen':inpgen_code,'fleur':fleur_code},'wf_parameters':wf_para,'fleur':fleur_code,'options':options}banddos_workchain=submit(FleurBandDosWorkChain,**inputs)