Fleur Magnetic Anisotropy Energy workflow
Current version: 0.2.0
Class:
FleurMaeWorkChainString to pass to the
WorkflowFactory():fleur.maeWorkflow type: Scientific workchain, force-theorem subgroup
Aim: Calculate Magnetic Anisotropy Energies along given spin quantization axes
Contents
Import Example:
from aiida_fleur.workflows.mae import FleurMaeWorkChain
#or
WorkflowFactory('fleur.mae')
Description/Purpose
This workchain calculates Magnetic Anisotropy Energy over a given set of spin-quantization axes. The force-theorem is employed which means the workchain converges a reference charge density first then it submits a single FleurCalculation with a <forceTheorem> tag.
The task of the workchain us to calculate the energy difference between two or several structures having a different magnetisation profile:

To do this, the workchain employs the force theorem approach:
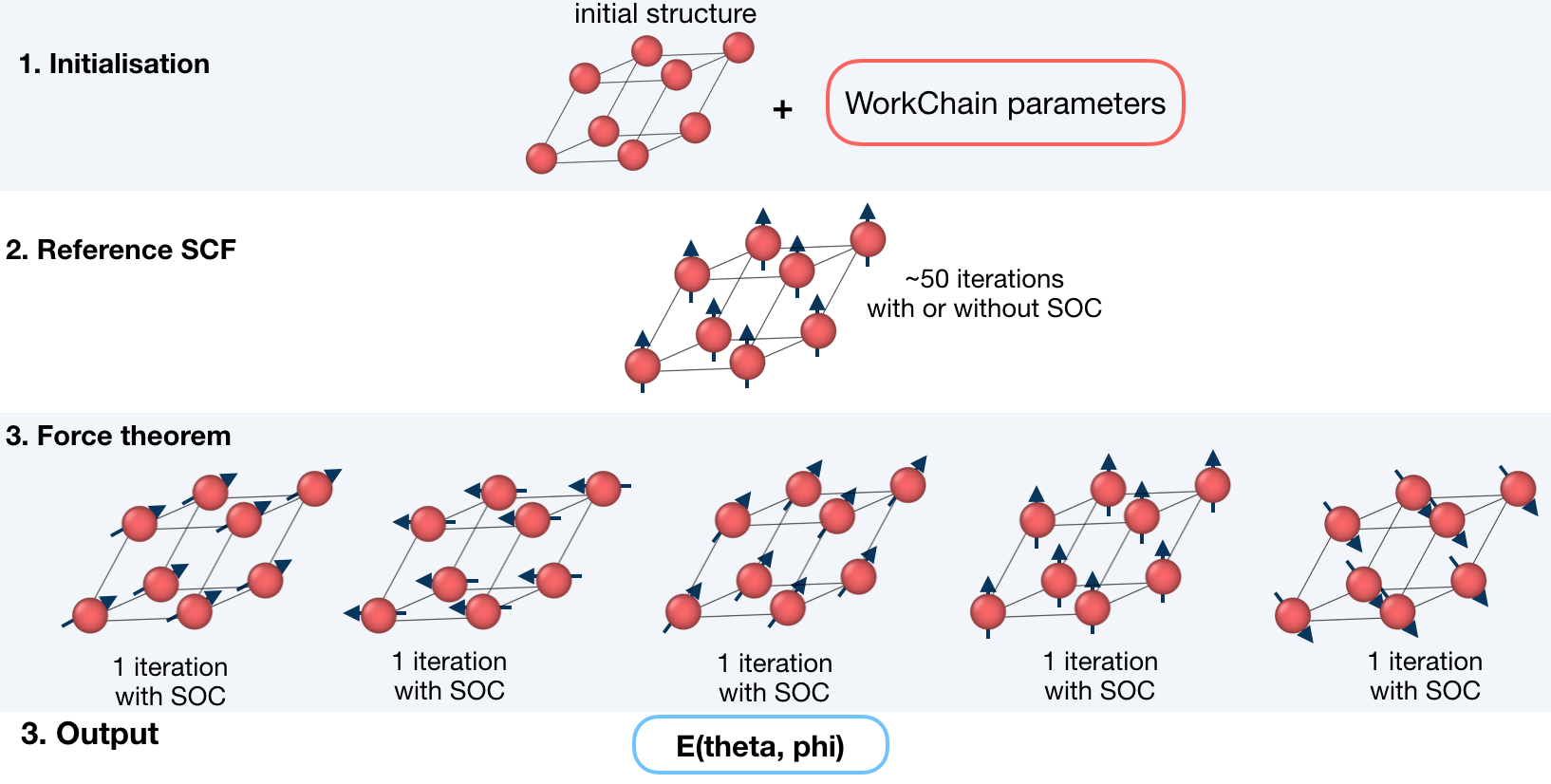
It is not always necessary to start with a structure. Setting up input parameters correctly (see Supported input configurations) one can start from a given FleuinpData, inp.xml or converged/not-fully-converged reference charge density.
Input nodes
The FleurMaeWorkChain employs
exposed feature of the AiiDA, thus inputs for the nested
SCF workchain should be passed in the namespace
scf.
name |
type |
description |
required |
|---|---|---|---|
scf |
namespace |
inputs for nested SCF WorkChain |
no |
fleur |
Fleur code |
yes |
|
wf_parameters |
Settings of the workchain |
no |
|
fleurinp |
no |
||
remote |
Remote folder of another calculation |
no |
|
options |
AiiDA options (computational resources) |
no |
Only fleur input is required. However, it does not mean that it is enough to specify fleur only. One must keep one of the supported input configurations described in the Supported input configurations section.
Workchain parameters and its defaults
wf_parameters
wf_parameters: Dict - Settings of the workflow behavior. All possible
keys and their defaults are listed below:
# -*- coding: utf-8 -*-
'sqa_ref': [0.7, 0.7], # sets theta and phi for the reference calc
'use_soc_ref': False, # True if reference calc should use SOC terms
'sqas_theta': [0.0, 1.57079, 1.57079], # a list of theta values for the FT
'sqas_phi': [0.0, 0.0, 1.57079], # a list of phi values for the FT
'add_comp_para': {
'only_even_MPI': False, # True if suppress parallelisation having odd number of MPI
'max_queue_nodes': 20, # Max number of nodes allowed (used by automatic error fix)
'max_queue_wallclock_sec': 86400 # Max number of walltime allowed (used by automatic error fix)
},
'soc_off': [], # a list of atom labels to switch off SOC term
'inpxml_changes': [] # additional changes before the FT step
soc_off is a python list containing atoms labels. SOC is switched off for species, corresponding to the atom with a given label.
Note
It can be that the specie correspond to several atoms and soc_off switches off SOC for atoms that was not intended to change. You must be careful and make sure that several atoms do not correspond to a given specie.
An example of soc_off work:
'soc_off': ['458']
changes
<species name="Ir-2" element="Ir" atomicNumber="77" coreStates="17" magMom=".00000000" flipSpin="T">
<mtSphere radius="2.52000000" gridPoints="747" logIncrement=".01800000"/>
<atomicCutoffs lmax="8" lnonsphr="6"/>
<energyParameters s="6" p="6" d="5" f="5"/>
<prodBasis lcutm="4" lcutwf="8" select="4 0 4 2"/>
<lo type="SCLO" l="1" n="5" eDeriv="0"/>
</species>
-----
<atomGroup species="Ir-2">
<filmPos label=" 458">1.000/4.000 1.000/2.000 11.4074000502</filmPos>
<force calculate="T" relaxXYZ="TTT"/>
<nocoParams l_relax="F" alpha=".00000000" beta=".00000000" b_cons_x=".00000000" b_cons_y=".00000000"/>
</atomGroup>
to:
<species name="Ir-2" element="Ir" atomicNumber="77" coreStates="17" magMom=".00000000" flipSpin="T">
<mtSphere radius="2.52000000" gridPoints="747" logIncrement=".01800000"/>
<atomicCutoffs lmax="8" lnonsphr="6"/>
<energyParameters s="6" p="6" d="5" f="5"/>
<prodBasis lcutm="4" lcutwf="8" select="4 0 4 2"/>
<special socscale="0.0"/>
<lo type="SCLO" l="1" n="5" eDeriv="0"/>
</species>
As you can see, I was careful about “Ir-2” specie and it contained a single atom with a label 458. Please also refer to Setting up atom labels section to learn how to set labels up.
sqas_theta and sqas_phi are python lists that set SOC theta and phi values.
sqa_ref sets a spin quantization axis [theta, phi] for the reference calculation if SOC terms are switched on by use_soc_ref.
options
options: Dict - AiiDA options (computational resources).
Example:
'resources': {"num_machines": 1, "num_mpiprocs_per_machine": 1},
'max_wallclock_seconds': 6*60*60,
'queue_name': '',
'custom_scheduler_commands': '',
'import_sys_environment': False,
'environment_variables': {}
Output nodes
out:Dict- Information of workflow results like success, last result node, list with convergence behavior"errors": [], "info": [], "initial_structure": "ac274613-27f5-4c0b-9d42-bae340007ab1", "is_it_force_theorem": true, "mae_units": "eV", "maes": [ 0.0006585155416697, 0.0048545112659747, 0.0 ], "phi": [ 0.0, 0.0, 1.57079 ], "theta": [ 0.0, 1.57079, 1.57079 ], "warnings": [], "workflow_name": "FleurMaeWorkChain", "workflow_version": "0.1.0"Resulting Magnetic Anisotropy Directions are sorted according to theirs theta and phi values i.e.
maes[N]corresponds totheta[N]andphi[N].
Supported input configurations
MAE workchain has several input combinations that implicitly define the workchain layout. Only scf, fleurinp and remote nodes control the behaviour, other input nodes are truly optional. Depending on the setup of the inputs, one of several supported scenarios will happen:
scf:
SCF workchain will be submitted to converge the reference charge density which will be followed be the force theorem calculation. Depending on the inputs given in the SCF namespace, SCF will start from the structure or FleurinpData or will continue converging from the given remote_data (see details in SCF WorkChain).
remote:
Files which belong to the remote will be used for the direct submission of the force theorem calculation.
inp.xmlfile will be converted to FleurinpData and charge density will be used as a reference charge density.remote + fleurinp:
Charge density which belongs to remote will be used as a reference charge density, however
inp.xmlfrom the remote will be ignored. Instead, the given fleurinp will be used. The aforementioned input files will be used for direct submission of the force theorem calculation.
Other combinations of the input nodes scf, fleurinp and remote are forbidden.
Warning
One must follow one of the supported input configurations. To protect a user from the workchain misbehaviour, an error will be thrown if one specifies e.g. both scf and remote inputs because in this case the intention of the user is not clear either he/she wants to converge a new charge density or use the given one.
Error handling
A list of implemented exit codes:
Code |
Meaning |
|---|---|
230 |
Invalid workchain parameters |
231 |
Invalid input configuration |
233 |
Input codes do not correspond to fleur or inpgen codes respectively. |
235 |
Input file modification failed. |
236 |
Input file was corrupted after modifications |
334 |
Reference calculation failed. |
335 |
Found no reference calculation remote repository. |
336 |
Force theorem calculation failed. |
Example usage
# -*- coding: utf-8 -*- from aiida.orm import load_node, Dict from aiida.engine import submit from aiida_fleur.workflows.mae import FleurMaeWorkChain structure = load_node(STRUCTURE_PK) fleur_code = load_node(FLEUR_PK) inpgen_code = load_node(INPGEN_PK) wf_para = Dict(dict={'sqa_ref': [0.7, 0.7], 'use_soc_ref': False, 'sqas_theta': [0.0, 1.57079, 1.57079], 'sqas_phi': [0.0, 0.0, 1.57079], 'add_comp_para': { 'only_even_MPI': False, 'max_queue_nodes': 20, 'max_queue_wallclock_sec': 86400 }, 'soc_off': [], 'inpxml_changes': [], }) options = Dict(dict={'resources': {'num_machines': 1, 'num_mpiprocs_per_machine': 24}, 'queue_name': 'devel', 'custom_scheduler_commands': '', 'max_wallclock_seconds': 60*60}) parameters = Dict(dict={'atom': {'element': 'Pt', 'lmax': 8 }, 'atom2': {'element': 'Fe', 'lmax': 8, }, 'comp': {'kmax': 3.8, }, 'kpt': {'div1': 20, 'div2': 24, 'div3': 1 }}) wf_para_scf = {'fleur_runmax': 2, 'itmax_per_run': 120, 'density_converged': 0.2, 'mode': 'density' } wf_para_scf = Dict(dict=wf_para_scf) options_scf = Dict(dict={'resources': {'num_machines': 2, 'num_mpiprocs_per_machine': 24}, 'queue_name': 'devel', 'custom_scheduler_commands': '', 'max_wallclock_seconds': 60*60}) inputs = {'scf': {'wf_parameters': wf_para_scf, 'structure': structure, 'calc_parameters': parameters, 'options': options_scf, 'inpgen': inpgen_code, 'fleur': fleur_code }, 'wf_parameters': wf_para, 'fleur': fleur_code, 'options': options } res = submit(FleurMaeWorkChain, **inputs)